

Was ist die Fabry Krankheit?
Fabry wurde erstmals 1898 unabhängig voneinander von zwei Ärzten beschrieben: dem Engländer Dr. William Anderson und dem Deutschen Dr. Johannes Fabry. Die Krankheit wird deshalb manchmal auch als Anderson Krankheit oder Anderson-Fabry Krankheit bezeichnet.
Fabry Krankheit ist eine seltene erbliche Krankheit, die schätzungsweise 1 von 40'000 bis 60'000 Männern betrifft. Die Krankheit tritt auch bei Frauen auf, obwohl die Prävalenz unbekannt ist. Diese Krankheit zählt zu den lysosomalen Speicherkrankheiten, die durch den Mangel an einem bestimmten Enzym gekennzeichnet sind.
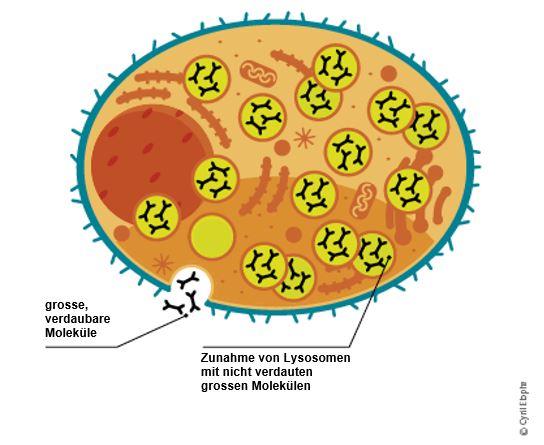
Patienten mit Fabry fehlt es an dem wichtigen Enzym α-Galaktosidase A, das vom Körper nicht oder nur in sehr geringen Mengen hergestellt wird. Ohne dieses Enzym werden bestimmte Stoffe oder Abfallprodukte nicht aus dem Körper entfernt, sondern können sich in den Zellen wichtiger Organe wie den Nieren und dem Herz sowie im zentralen Nervensystem anreichern. Diese allmähliche Ansammlung (= lysosomale Speicherung) von Abfallstoffen stört die normale Funktionsweise der Zellen und verursacht Fabry.
Über das Fabry-Enzym (α-Galaktosidase A)
α-Galaktosidase A ist nur eines von mehreren Enzymen, die normalerweise in den Lysosomen vorhanden sind. Die Lysosomen sind kleine «Recyclingzentren» in unseren Zellen, die eine wichtige Rolle dabei spielen, Abfallstoffe in kleinere Einheiten aufzuspalten, die von der Zelle wiederverwertet oder aus dem Körper ausgeschieden werden können.
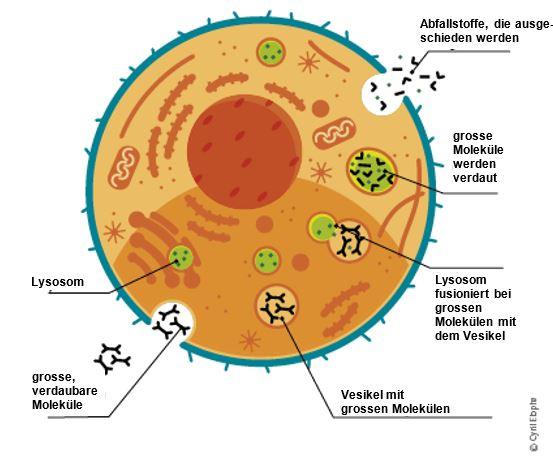
Ein Mangel an α-Galaktosidase A führt zur Anhäufung von fetthaltigen Abfallstoffen (Lipiden) in den Zellen. Das wichtigste dieser Abfallprodukte ist Globotriaosylzeramid (GL-3).
MAT-CH-2000186-6.0 - 01/2024
Drucke diese Seite