

Qu'est-ce que la maladie de Fabry?
La maladie de Fabry a été décrite pour la première fois en 1898 par deux médecins, indépendamment l'un de l'autre: l'Anglais Dr William Anderson et l'Allemand Dr Johannes Fabry. La maladie est donc également parfois appelée maladie d'Anderson ou maladie d'Anderson-Fabry.
La maladie de Fabry est une maladie héréditaire rare qui touche environ 1 homme sur 40 000 à 60 000. La maladie se manifeste également chez les femmes, mais sa prévalence est inconnue.
Cette maladie fait partie des maladies de surcharge lysosomale, caractérisées par le déficit d'une enzyme spécifique.
Chez les patients atteints de la maladie de Fabry, ce déficit concerne l'enzyme essentielle α-galactosidase A, qui n'est pas ou que très peu produite par l'organisme. Sans cette enzyme, certaines substances ou résidus ne sont pas éliminés de l'organisme, et peuvent s'accumuler dans les cellules d'organes essentiels tels que les reins et le cœur ou encore le système nerveux central. Cette accumulation progressive (= surcharge lysosomale) de résidus perturbe le fonctionnement normal des cellules et est à l'origine de la maladie de Fabry.
À propos de l'enzyme de Fabry (α-galactosidase A)
L'α-galactosidase A n'est qu'une des différentes enzymes qui sont normalement présentes dans les lysosomes. Les lysosomes sont de petits «centres de recyclage» au sein de nos cellules, qui jouent un rôle essentiel dans la décomposition des substances résiduelles en petites unités qui sont alors recyclées par la cellule ou bien éliminées par l'organisme.
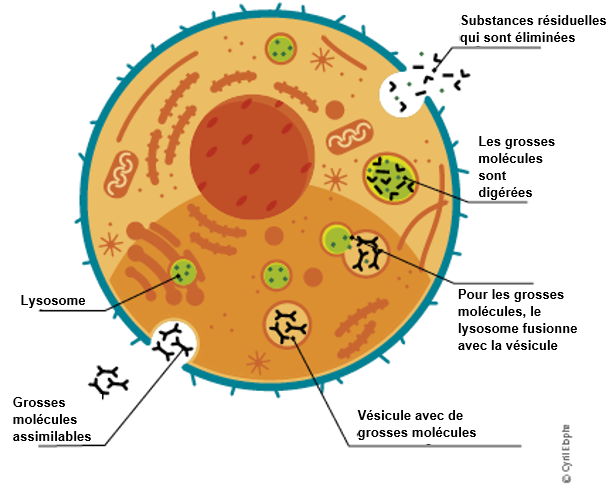
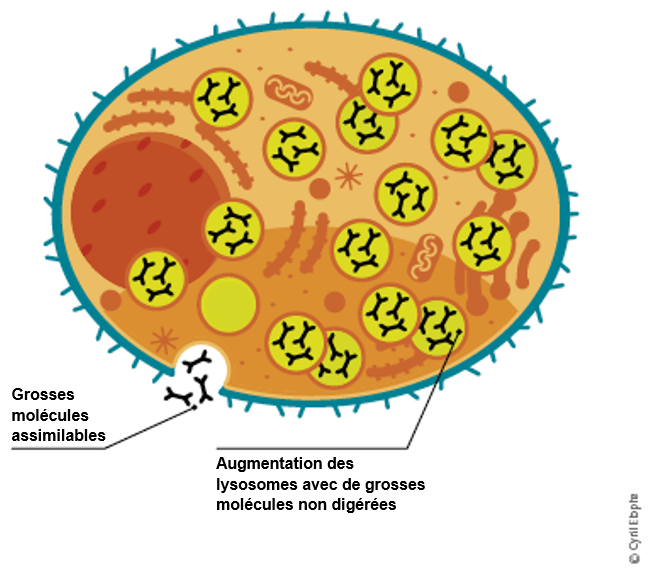
Un déficit en α-galactosidase A entraîne une accumulation de substances grasses résiduelles (lipides) dans les cellules. La plus importante de ces substances résiduelles est la globotriaosylcéramide (GL-3).
MAT-CH-2000187-5.0 - 01/2024
Imprimer cette page