

Che cos'è la malattia di Fabry?
La malattia di Fabry fu scoperta nel 1898 da due medici, in modo indipendente l'uno dall'altro: il medico inglese dott. William Anderson e il medico tedesco dott. Johannes Fabry. Per questo motivo, talvolta la malattia viene anche detta malattia di Anderson o malattia di Anderson-Fabry.
La malattia di Fabry è una malattia rara ereditaria che colpisce circa 1 su 40'000 - 60'000 uomini. La malattia si verifica anche nelle donne, anche se la prevalenza è sconosciuta. Questa malattia appartiene alle malattie da accumulo lisosomiale, caratterizzate dalla carenza di un enzima specifico.
I pazienti affetti da malattia di Fabry non possiedono l'importante enzima α-galattosidasi A, che non viene prodotto dall'organismo o viene prodotto solo in quantità molto limitate. Senza questo enzima, l'organismo non riesce a smaltire determinate sostanze o scorie che possono quindi accumularsi nelle cellule di organi importanti come i reni, il cuore e il sistema nervoso centrale. Questo graduale accumulo (=accumulo lisosomiale) di scorie compromette la normale funzionalità cellulare e causa l'insorgenza della malattia di Fabry.
Informazioni sull'enzima di Fabry (α-galattosidasi A)
L'α-galattosidasi A è solamente uno dei numerosi enzimi generalmente presenti all'interno dei lisosomi. I lisosomi sono dei piccoli "centri di riciclaggio" nella cellula e rivestono un ruolo importante nella scomposizione delle scorie in unità più piccole che possono essere riciclate dalla cellula oppure espulse dall'organismo.
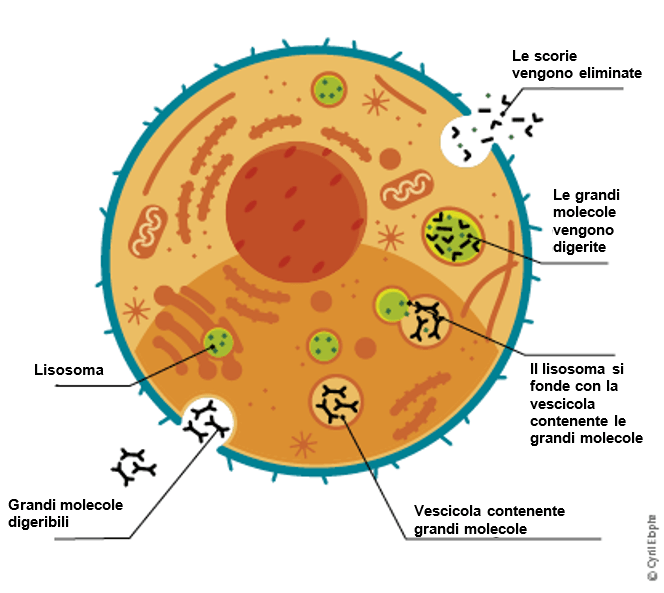
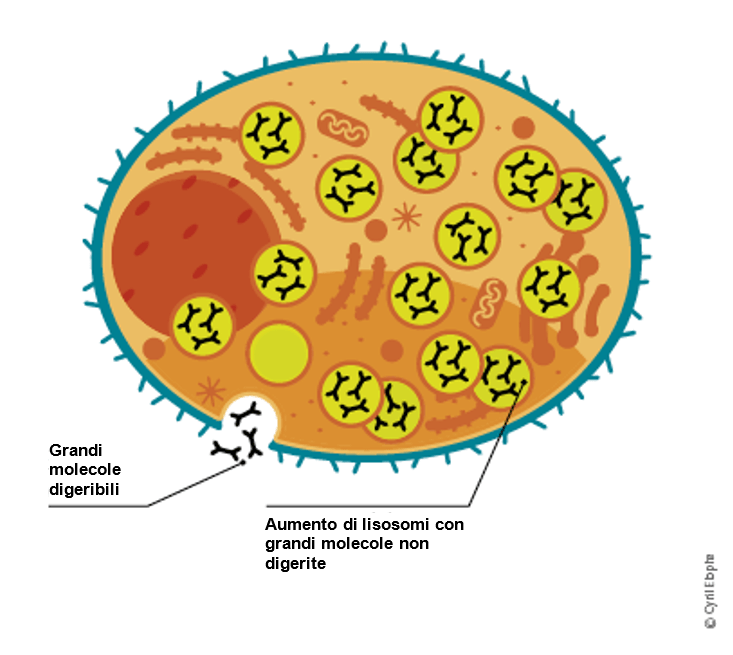
Una carenza di α-galattosidasi A comporta l'accumulo di scorie (lipidi) nelle cellule. La più importante di queste scorie è la globotriaosilceramide (GL-3).
MAT-CH-2100313-5.0 - 01/2024
Stampa questa pagina